Antibody drug conjugates (ADCs) are targeted cancer therapies that combine the specificity of monoclonal antibodies with the potency of cytotoxic drugs. In contrast to conventional chemotherapies, which do not discriminate between healthy and cancerous cells, ADCs are designed to deliver cytotoxic drugs directly to tumors and are therefore considered to be a more efficacious and safer therapeutic option. In this article, we comment on the progress as well as the safety and efficacy challenges associated with ADC development.
Firstly, how do ADCs work?
ADCs (have been) described as the magic bullets of cancer treatment, because they combine the specific targeting ability of monoclonal antibodies with the potency of small molecule cytotoxic drugs. Each ADC comprises 3 components:
- A monoclonal antibody (mAb)
- a stable linker
- a payload (e.g. a cytotoxic drug)
When an ADC is administered, the mAb binds a tumor antigen that is preferentially expressed on the tumor cell surface. This leads to the formation of an ADC-antigen complex. Covalent linkage between the mAb and the cytotoxic drug prevents premature release of the drug into the bloodstream. Once the mAb has bound its target, the ADC-antigen complex is taken up by the cells via receptor-mediated endocytosis. The cytotoxic drug is then released, where it typically targets DNA replication and/or DNA repair, microtubules or topoisomerase activity, resulting in cytotoxicity and apoptosis of the tumor cells.
As a minimum, the following criteria must be met for an ADC to be safe and efficacious
- The mAb must selectively bind the tumor-specific target antigen.
- The ADC-antigen complex must be internalized.
- The antibody must become degraded by lysosomes inside the target cell.
- The drug payload must be released within the cytoplasm of the target cell AND interact with its intracellular target.
- It is critical to control the drug to antibody ratio in ADC development to avoid the so-called bystander effect, whereby a small amount of the drug is unintentionally released in the extracellular environment where it is taken up by neighboring cells and causes toxicity.
ADC approvals to date:
In the four decades since ADCs first emerged (1), many advances in hybridoma and site-specific conjugation technology have yielded newer generations that comprise more highly-specific mAbs, novel and better linkers, improvements in drug to antibody ratios, and show superior chemical consistency. In addition, continued diversification within the mAb targets and cytotoxic payloads is expected to further broaden the scope of cancers that can be treated with ADC therapy.
As of October 2023, 13 of the 15 ADCs approved to date remain on the market for the treatment of various blood and solid cancers: most of these target HER2+ cancers by incorporating the unconjugated HER2-targeting mAb trastuzumab in their backbone (Table 1).
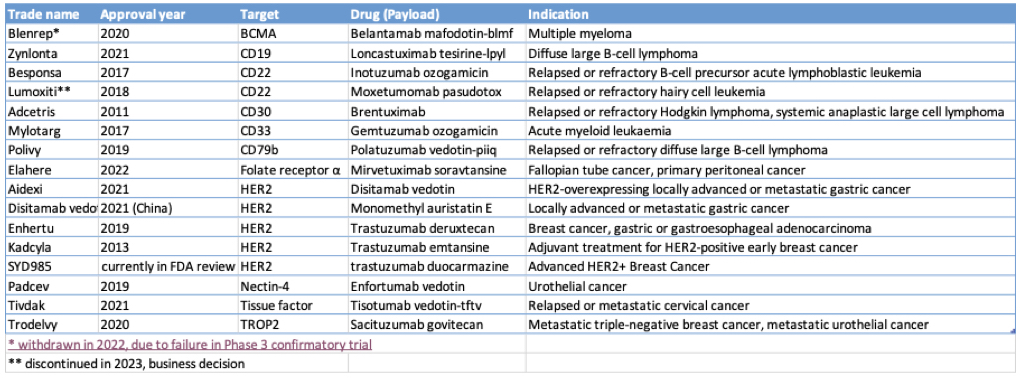
Table 1: ADCs
Despite progress in the field, far too many ADCs are failing clinical trials!
While the list of ADC approvals so far and the 150+ ongoing clinical trials for new ADCs add to hopes about the vast potential of ADCs to treat cancer, a striking number of ADCs fail clinical trials.
Representative examples of more than 90 ADCs discontinued following clinical testing include vadastuximab talirine developed by Genentech and discontinued in 2017 following fatal infections in Phase 3 clinical trial participants, MEDI4276 developed by AstraZeneca/MedImmune and discontinued at Phase 1 due to payload-related toxicities and limited efficacy at the maximum tolerable dose (2), and Rovalpituzumab Tesirine developed by AbbVie and discontinued in 2019 after Phase 3 trials showed no survival benefit compared to control subjects.
According to a detailed literature review of the ADC clinical landscape published in August 2023, the prevailing reason for the discontinuation of 92 ADC trials to date is minimal or insufficient efficacy at the tolerated doses; in other words, the candidates have not been safe enough to be dosed at the levels required for efficacy (3).
Analysis of the clinical data from ADC trials has revealed that dose-limiting toxicities are often shared by different ADCs that deliver the same cytotoxic payload, and this occurs independently of the target antigen or cancer type. In general, dose-limiting toxicities are off-target, i.e. they occur in cells and tissues that don’t express the targeted antigen; this limits the tolerable ADC dosage to levels that provide little or no efficacy resulting in clinical failure as mentioned above (4).
De-risking ADC development requires rigorous pre-clinical testing in appropriate models
While the sources of ADC toxicity have been covered in a recent literature review (4), and will not be discussed in detail here. It is worth noting that the individual components of any ADC can impact its overall efficacy and safety profile, and it is imperative during the pre-clinical stages to dissect which ADC design feature or component is driving the desired efficacy or undesired toxicity.
Many of the toxicity and efficacy hurdles faced in ADC clinical trials could likely be mitigated by rigorous pre-clinical testing in models that more accurately represent the human target cell or tissue type in the relevant human disease context. Some of the major problems associated with current pre-clinical efficacy and toxicity models include:
- Rodent models are inadequate models for toxicity and efficacy. Despite genetic and biological similarities to humans, rodent models do not provide reliable readouts for efficacy or toxicity associated with experimental ADCs because of a lack of antibody cross-reactivity to the target.
- Risky assumptions about non-human primates. The cynomolgus monkey has become the most widely used non-human primate (NHP) in pre-clinical toxicology analyses, based on the assumption that it responds to drugs similarly to humans given the physiological and genetic similarities between the two species. However, in reality, the existing phenotypic and genotypic differences between the two can result in drugs appearing to be safe in NHPs but actually lethal in humans once evaluated in clinical trials. This limitation of using NHPs is exacerbated by the fact that some research NHPs are lab-born while others are sourced from the wild, leading to variation that may skew inter- and intra-experimental comparisons.
- Major differences in the expression of immunological markers between species. For example, the Fcγ receptor CD16, which is critical for NK cell proliferation and degranulation, is expressed on granulocytes in humans and sooty mangabeys, but not in macaques or baboons, while CD56 is found on monocytes in macaques but is a canonical natural killer cell marker in humans (5). Although not an ADC, the clinical failure of TeGenero’s anti-CD28 mAb candidate (Fialuridine) is an important example of the potential disastrous consequences of differential CD expression between humans and NHPs when testing biological drugs. When administered to humans at a fraction of the dose found to be safe in four animal species including cynomolgus monkeys, all trial participants became rapidly ill due to a near fatal cytokine storm that was unforeseen from pre-clinical animal studies due to differences in CD28 expression between species (6, and references therein).
- Challenges in understanding ADC pharmacokinetics. ADC pharmacokinetics are characterized by a continuous decrease in the concentration of structurally intact ADC and naked antibody as the ADC is internalized and the antibody is cleared. Among the major factors that govern antibody clearance are the mononuclear phagocyte system and Fc receptor (FcRn)-mediated recycling. Differences between the human immune system and that of the animal models used can lead to inaccurate pre-clinical predictions about how an experimental ADC will interact with a human body.
That was it for our introduction to ADCs. Stay tuned for a future article where we discuss emerging cell-based models for efficacy and toxicity that can address the challenges described above!
References:
- Ford CH, Newman CE, Johnson JR, Woodhouse CS, Reeder TA, Rowland GF, Simmonds RG. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br J Cancer. 1983 Jan;47(1):35-42.
- Pegram MD, Hamilton EP, Tan AR, Storniolo AM, Balic K, Rosenbaum AI, Liang M, He P, Marshall S, Scheuber A, Das M, Patel MR. First-in-Human, Phase 1 Dose-Escalation Study of Biparatopic Anti-HER2 Antibody-Drug Conjugate MEDI4276 in Patients with HER2-positive Advanced Breast or Gastric Cancer. Mol Cancer Ther. 2021 Aug;20(8):1442-1453.
- Maecker H, Jonnalagadda V, Bhakta S, Jammalamadaka V, Junutula JR. Exploration of the antibody-drug conjugate clinical landscape. MAbs. 2023 Jan-Dec;15(1):2229101.
- Nguyen TD, Bordeau BM, Balthasar JP. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers (Basel). 2023 Jan 24;15(3):713. doi: 10.3390/cancers15030713.
- Carter DL, Shieh TM, Blosser RL, Chadwick KR, Margolick JB, Hildreth JE, Clements JE, Zink MC. CD56 identifies monocytes and not natural killer cells in rhesus macaques. Cytometry. 1999 Sep 1;37(1):41-50.
- Eastwood D, Findlay L, Poole S, Bird C, Wadhwa M, Moore M, Burns C, Thorpe R, Stebbings R. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br J Pharmacol. 2010 Oct;161(3):512-26. doi: 10.1111/j.1476-5381.2010.00922.x.
Further reading: Timeline: charting the choppy history of ‘magic bullet’ antibody-drug conjugates (Pharmaceutical Technology, 2021).
Karen O’Hanlon Cohrt is an independent Science Writer with a PhD in biotechnology from Maynooth University, Ireland (2011). After her PhD, Karen relocated to Denmark where she held postdoctoral positions in mycology and later in human cell cycle regulation, before moving to the world of drug discovery. Karen has been a full-time science writer since 2017, and has since then held numerous contract roles in science communication and editing spanning diverse topics including diagnostics, molecular biology, and gene therapy. Her broad research background provides the technical know-how to support scientists in diverse areas, and this in combination with her passion for learning helps her to keep abreast of exciting research developments as they unfold. Karen is currently based in Ireland, and you can follow her on Linkedin here.